
Module 3 lab index | Next lab day >
Introduction
Today we will continue the discussion that we began in lecture about cell-biomaterial interactions and cartilage tissue engineering, with the ultimate goal of designing an experiment probing chondrocyte phenotype development and/or maintenance. You will also get some practice with cell culture today, to prepare you for beginning your experiment next time.
These papers on chondrocyte tissue culture and cartilage tissue engineering will give you a sense of some design options. You are also welcome to search the scientific literature on your own for further ideas.
Brodkin, K. R., A. J. Garcia, and M. E. Levenston. "Chondrocyte phenotypes on different extracellular matrix monolayers." Biomaterials 25 (2004): 5929-5938.
Kong, H. Y., et al. "Controlling Rigidity and Degradation of Alginate Hydrogels via Molecular Weight Distribution." Biomacromolecules 5 (2004): 1720-1727.
Heywood, H. K., et al. "Cellular Utilization Determines Viability and Matrix Distribution Profiles in Chondrocyte-Seeded Alginate Constructs." Tissue Engineering 10, no. 9/10 (2004): 1467-1479.
Genes, N. G., et al. "Effect of substrate mechanics on chondrocyte adhesion to modified alginate surfaces." Archives of Biochemistry and Biophysics 422 (2004): 161-167.
Domm, C., et al. "Influence of Various Alginate Brands on the Redifferentiation of Dedifferentiated Bovine Articular Chondrocytes in Alginate Bead Culture under High and Low Oxygen Tension." Tissue Engineering 10, no. 11/12 (2004): 1796-1805.
Lee, C. S. D., et al. "Integration of layered chondrocyte-seeded alginate hydrogel scaffolds." Biomaterials 28 (2007): 2987-2993.
Yoon, D. M., et al. "Addition of Hyaluronic Acid to Alginate Embedded Chondrocytes Interferes with Insulin-like Growth Factor-1 Signaling In Vitro and In Vivo." Tissue Engineering Part A 15, no. 11 (2009): 3449-3459.
Bosnakovski, D., et al. "Chondrogenic Differentiation of Bovine Bone Marrow Mesenchymal Stem Cells (MSCs) in Different Hydrogels: Influence of Collagen Type II Extracellular Matrix on MSC Chondrogenesis." Biotechnology and Bioengineering 93, no. 6 (2006): 1152-1163.
Protocols
Half the class today will start in the cell culture facility and half will start with experimental design. Midway through class, you'll switch places. In your notebooks, you should only write up Part 2 of the protocol. For Part 1, the only thing you need to write down is your cell count data, for use in a later FNT calculation. The Part 1 protocol will be posted on each tissue culture hood for your reference.
Part 1: Practice Cell Culture
Background
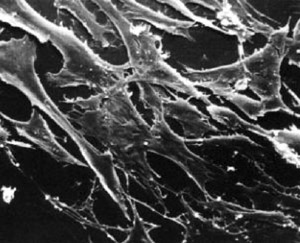
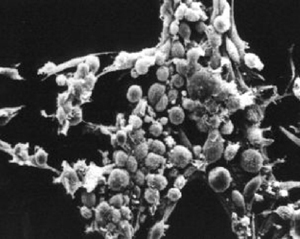
Normal and transformed mouse fibroblasts. (Courtesy of G. Steven Martin. Used with permission.)
In the past century, we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. Tissue culture was originally developed about 100 years ago as a method for learning about mammalian biology. The term tissue culture was originally coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using cells rather than tissues. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.
What types of cells do people study, and where do they come from? Cells that come from a tissue are called primary cells, because they come directly from an animal. It is very difficult to culture primary cells, largely because primary cells that are placed in culture divide only a limited number of times. This limitation in the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to constantly remove tissues from animals in order to complete a study. Cell isolation processes can be quite labour-intensive, and also can complicate data analysis due to inherent animal-to-animal variation. To get around this problem, people have studied cells that are immortal, which means that they can divide indefinitely. Some inherent cell-to-cell variation still exists in such cells. Moreover, the genetic changes that cause immortality may affect experimental outcomes.
One type of familiar immortalized cell is the cancer cell. Tumor cells continuously divide allowing cancer to invade tissues and proliferate. Cancer cells behave the same way in culture, and under the right conditions, cells can be taken from a tumor and divide indefinitely in culture. Another type of immortalized cell is the embryonic stem cell. Embryonic stem cells are derived from an early stage embryo, and these cells are completely undifferentiated and pluripotent, which means that under the right conditions, they can become any mammalian cell type. Mouse embryonic stem cells have become a valuable research tool, and it is this cell type that we will be using for our practice cell culture today.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free (and clotting-factor free) component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cells doubles about once per day whereas many common bacteria can double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
One major objective for this experimental module is for you to learn how to perform tissue culture. Today you will learn how to get mouse embryonic stem cells to grow in a dish and also how to prevent contaminants from getting into your cell cultures. Next time you will set up 3D cultures of bovine-derived cells in alginate beads.
Protocol
Each of you will have a 35 mm dish of mouse embryonic stem (MES) cells that you will use to seed a six-well dish. You and your partner will seed the dishes at different concentrations so you should decide who will seed at 1:10 and who will seed at 1:2. We will begin with a brief demo about sterile technique how to use the tissue culture hoods.
- Each tissue culture hood is partly set up for you. Finish preparing your hood according to the demo, first bringing in any remaining equipment you will need, then picking up the pre-warmed reagents from the water bath. Don't forget to spray everything down with 70% ethanol.
- One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Aspirate the media from the cells using a sterile Pasteur pipet. Dip the pipet in your beaker of ethanol when needed (to clean it).
- Wash the cells by adding 2 ml PBS using a 5 mL pipet. Slightly tip the dish back and forth to rinse all the cells, and then aspirate the liquid.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Using a 2 ml pipet, add 0.7 ml of trypsin to the flask. Be careful not to pull up liquid too quickly or it will go all the way up your pipet into the pipet-aid!
- Tip the flask in each direction to distribute the liquid evenly. Incubate the cells at 37°C for 3-5 minutes, until the cells round up and are easily dislodged from the plate by tapping.
- While you are waiting, you can aspirate the gelatin from the six-well dish in your hood (one dish per pair).
- The teaching faculty previously added 1 mL of sterile 0.1% gelatin to the two leftmost wells of the dish. MES must grow on either a "feeder layer" of fibroblasts, or on a gelatin-coated dish. The pre-treatment must be done for at least 10 minutes.
- After retrieving your cells, add 1.3 ml of media to the trypsinized MES cells and pipet the liquid up and down "triturate") to remove the cells from the plastic and suspend them in the liquid. Remove a small amount of the suspension (perhaps 50 μL) to an eppendorf tube.
- According to the procedures below, either begin counting your cells or begin plating them (no matter what count you get, you will plate a 1:2 and a 1:10 dilution), depending on microscope availability.
- Take your cell aliquot to the inverted microscopes and fill one chamber of a hemocytometer with 10 μL of the cell suspension.
- This slide has an etched grid of nine large squares. The square in the center is further etched into 25 squares each with a volume of 0.1 ul and 16 tiny chambers (4x4 pattern). The concentration of cells in a sample can be determined by counting the cells that fall within the 4x4 pattern and then multiplying by 10,000 to determine the number of cells/ml.
- You should count the cells in the four corner squares of the 25 square grid, then average the numbers to determine the concentration of cells in your suspension. Save your raw data for the Day 2 FNT assignment!
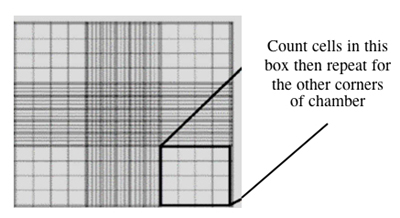
Counting cells using a hemocytometer
- You and your partner will seed at different concentrations. Decide if you will try the 1:10 or 1:2 dilution and add the appropriate amount of cell suspension to 3 ml of media in a 15 ml conical tube.
- Note that the dilution factor refers to the volume of the original cell culture, not the volume that you are moving the cells into. (If the surface area of the 35 mm dish and 6-well dish were very different, we would also want to take that into account.)
- Remove the gelatin from the six-well dish (if you haven't already) and add 3 ml of your cell dilution to one of the gelatin-treated wells. Your partner will use the other treated well in the same dish. Be sure to label your plate, then return your cells to the incubator.
- Label the plate lid with your group colour, today's date, and the cell line (called "J1"). Label the well you used with your initials and the cell dilution you did, and make sure your partner does the same.
- Aspirate any remaining cell suspensions to destroy them and clean up the hood. Dispose any vessels that held cells in the biohazard waste and any sharps in the biohazard bins. The next group who uses your hood should find the surfaces wiped down, no equipment that you brought in left inside, and the sash closed. Do leave the equipment that was already there.
Part 2: Experiment Design
The overall goal of this module is to test the effect of the surrounding environment on cell phenotype. In particular, you will work with primary chondrocytes and/or mesenchymal stem cells in 3D gel culture. The specific aspects of phenotype assayed will be collagen I and collagen II transcript and protein levels (these are markers for cell type), as well as the general cell characteristic of viability. You are free to propose one other assay if you wish, as you will likely have some extra time in the latter part of the module. One possibility is a DMMB colorimetric assay for proteoglycan content. You will be able to compare some of the data in your 3D culture experiments with control data from freshly isolated chondrocytes and stem cells.
Each pair of you will test two samples. Both samples will be grown in 3D alginate bead culture, and should have one parameter varied between them. For example, you might try changing the mechanical properties of the beads (how would you do this?), or the cell density within the beads. Be as creative as you like! If your protocol requires a new reagent or equipment to be ordered, we will do our best to get it in time. Of course, two samples is not very many for determining a trend. You are more than welcome to join up with another group or two in order to expand the range of the parameter you are testing (e.g., testing four cell densities instead of two). If everyone wants to test something different, that's okay too.
Most of you should explore conditions for maintaining or destroying chondrocyte phenotype - recall from lecture that chondrocytes grown without proper signals, for example in simple monolayer culture, tend to de-differentiate to a fibroblastic phenotype over time. We will also have some mesenchymal stem cells for a few groups to work with, and investigate conditions that promote chondrogenesis. Please see the teaching faculty with your proposed experiment, as we have a limited amount of each cell type.
Ultimately, you should hand in the following information:
- Type of alginate to be used, and at what %
- Cell type to be used
- Cell density per condition (in cells/mL)
- Total number of cells needed
- Unique medium formulations or supplements to be used
- Unique systems (mechanical, electrical, etc.) to be used
Per 3D sample, you will prepare 1 mL of alginate beads (thus a cell density of 5 million cells per mL would require 5 million cells per sample, 10 million total). As discussed in the pre-lab lecture, you should also write a brief summary in your notebook of three of the eight papers that you read today.
Materials available for 3D culture
| ALGINATE COMPANY | ALGINATE NAME | VISCOSITY | G/M RATIO |
|---|---|---|---|
| Sigma Aldrich | "low viscosity" | 250 cps at 2% | "high M" |
| FMC Biopolymer | Protanal LF 120M | 70-150 cps at 1% | ~40/60 |
| FMC Biopolymer | Protanal LF 10/60 | 20-70 cps at 1% | ~70/30 |
FMC Biopolymer alginates are samples generously donated by the company.
Collagen I and II gels are also available upon request. Keep in mind that using collagen directly will confound your protein assay results (unless you devise some controls), but not the transcript-level assay results.
Standard Stem Cell Medium
- Expansion Medium
- Low-glucose DMEM
- 10% FCS
- Penicillin/Streptomycin
- Amphotericin B
- HEPES buffer, 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- up to 5 ng/mL bFGF (basic fibroblast growth factor)
- Differentiation Medium
- Hi-glucose DMEM
- FCS and/or ITS+1 (insulin/transferrin/selenium)
- Penicillin/Streptomycin
- Amphotericin B
- Non-essential amino acids
- Sodium pyruvate
- Proline (400 μM)
- HEPES (10 mM)
- Chondrogenic factors
- TGF-beta1 (10 ng/mL)
- Dexamethasone (100 nM)
- Ascorbate (40 μg/mL)
Standard Chondrocyte Medium
- Growth medium
- Hi-glucose DMEM
- 10% FCS (or 2% FCS with ITS, for more defined media)
- Penicillin/Streptomycin
- Amphotericin B
- Non-essential amino acids
- Sodium pyruvate
- Proline (400 μM)
- HEPES (10 mM)
- Ascorbate(20 μg/mL)
For Next Time
- Familiarize yourself with the cell culture portion of Day 2 of this module. The better prepared we all are, the less likely it is that the day will run long. The hoods will be set up for you when you come in.
- By the end of class on Day 2, write a two or three sentence description of your design plan and expected assay results, and post it for the rest of the class. (Assay result expectations should be stated in a relative fashion: e.g., "we think [3D sample 1] will maintain a chondrocyte-like phenotype better than [3D sample 2], because..." You might also comment on cell viability, if you expect it to vary among your samples.) This posting will count for homework credit.